$ editconf -f conf.gro -o complex_newbox.gro -c -bt dodecahedron -d 1.0
Now that we have defined a box, we can fill it with solvent (water). Solvation is accomplished using genbox:
$ genbox -cp complex_newbox.gro -cs spc216.gro -o complex_solv.gro -p topol.top
What genbox has done is keep track of how many water molecules it has added, which it then writes to your topology to reflect the changes that have been made. Note that if you use any other (non-water) solvent, genbox will not make these changes to your topology! Its compatibility with updating water molecules is hard-coded
We now have a solvated system that contains a charged protein. The output of pdb2gmx told us that the protein has a net charge of -35e (based on its amino acid composition). If you missed this information in the pdb2gmx output, look at the last line of your [ atoms ] directive in topol.top; it should read (in part) "qtot 6." Since life does not exist at a net charge, we must add ions to our system.
The tool for adding ions within GROMACS is called genion. What genion does is read through the topology and replace water molecules with the ions that the user specifies. The input is called a run input file, which has an extension of .tpr; this file is produced by the GROMACS tool grompp (GROMACS pre-processor), which will also be used later when we run our first simulation. What grompp does is process the coordinate file and topology (which describes the molecules) to generate an atomic-level input (.tpr). The .tpr file contains all the parameters for all of the atoms in the system.
Use grompp to assemble a .tpr file, using any .mdp file. I use an .mdp file for running energy minimization, since they require the fewest parameters and are thus the easiest to maintain. To produce a .tpr file with grompp, we will need an additional input file, with the extension .mdp (molecular dynamics parameter file); grompp will assemble the parameters specified in the .mdp file with the coordinates and topology information to generate a .tpr file.
An .mdp file is normally used to run energy minimization or an MD simulation, but in this case is simply used to generate an atomic description of the system. An example .mdp file (the one we will use) can be downloaded
here.
In reality, the .mdp file used at this step can contain any legitimate combination of parameters. I typically use an energy-minimization script, because they are very basic and do not involve any complicated parameter combinations.
Assemble your .tpr file with the following:
$ grompp -f ions.mdp -c complex_solv.gro -p topol.top -o ions.tpr
Now we have an atomic-level description of our system in the binary file ions.tpr. We will pass this file to genion:
$ genion -s ions.tpr -o complex_solv_ions.gro -p topol.top -pname NA -nname CL -np 35
When prompted, choose group 14 "SOL" (for this case) for embedding ions. You do not want to replace parts of your protein with ions.
In the genion command, we provide the structure/state file (-s) as input, generate a .gro file as output (-o), process the topology (-p) to reflect the removal of water molecules and addition of ions, define positive and negative ion names (-pname and -nname, respectively), and tell genion to add only the ions necessary to neutralize the net charge on the protein by adding the correct number of negative ions (-nn 8). You could also use genion to add a specified concentration of ions in addition to simply neutralizing the system by specifying the -neutral and -conc options in conjunction. Refer to the genion man page for information on how to use these options.
The names of the ions specified with -pname and -nname were force field-specific in previous versions of GROMACS, but have been standardized as of version 4.5. The specified ion names are always the elemental symbol in all capital letters, which is the [ moleculetype ] name that is then written to the topology. Residue or atom names may or may not append the sign of the charge (+/-), depending on the force field. Do not use atom or residue names in the genion command, or you will encounter errors in subsequent steps.
The solvated, electroneutral system is now assembled. Before we can begin dynamics, we must ensure that the system has no steric clashes or inappropriate geometry. The structure is relaxed through a process called energy minimization (EM).
The process for EM is much like the addition of ions. We are once again going to use grompp to assemble the structure, topology, and simulation parameters into a binary input file (.tpr), but this time, instead of passing the .tpr to genion, we will run the energy minimization through the GROMACS MD engine, mdrun.
Assemble the binary input using grompp using
this input parameter file:
grompp -f minim.mdp -c complex_solv_ions.gro -p topol.top -o em.tpr
Make sure you have been updating your topol.top file when running genbox and genion, or else you will get lots of nasty error messages ("number of coordinates in coordinate file does not match topology," etc).
We are now ready to invoke mdrun to carry out the EM:
mdrun -v -deffnm em
The -v flag is for the impatient: it makes mdrun verbose, such that it prints its progress to the screen at every step. The -deffnm flag will define the file names of the input and output. So, if you did not name your grompp output "em.tpr," you will have to explicitly specify its name with the mdrun -s flag. In our case, we will get the following files:
- em.log: ASCII-text log file of the EM process
- em.edr: Binary energy file
- em.trr: Binary full-precision trajectory
- em.gro: Energy-minimized structure
writing lowest energy coordinates.
Steepest Descents converged to Fmax < 1000 in 1671 steps
Potential Energy = -1.4910198e+06
Maximum force = 8.8282404e+02 on atom 123
Norm of force = 1.2215010e+01
There are two very important factors to evaluate to determine if EM was successful. The first is the potential energy (printed at the end of the EM process, even without -v). Epot should be negative, and (for a simple protein in water) on the order of 105-106, depending on the system size and number of water molecules. The second important feature is the maximum force, Fmax, the target for which was set in minim.mdp - "emtol = 1000.0" - indicating a target Fmax of no greater than 1000 kJ mol-1 nm-1. It is possible to arrive at a reasonable Epot with Fmax > emtol. If this happens, your system may not be stable enough for simulation. Evaluate why it may be happening, and perhaps change your minimization parameters (integrator, emstep, etc).
Let's do a bit of analysis. The em.edr file contains all of the energy terms that GROMACS collects during EM. You can analyze any .edr file using the GROMACS tools g_energy:
g_energy -f em.edr -o potential.xvg
At the prompt, type "10 0" to select Potential (10); zero (0) terminates input. You will be shown the average of Epot, and a file called "potential.xvg" will be written. To plot this data, you will need the Xmgrace plotting tool. The resulting plot should look something like this, demonstrating the nice, steady convergence of Epot:
Statistics over 1670 steps [ 1.0000 through 1670.0000 ps ], 1 data sets
All statistics are over 1322 points (frames)
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Potential -1.44656e+06 24000 71431.2 -155083 (kJ/mol)
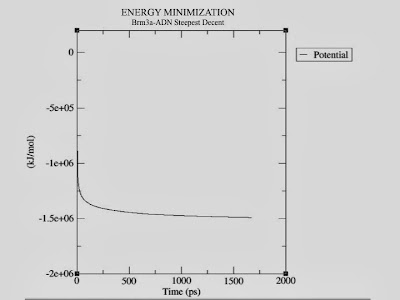 |
| Energy Minimization plot |
Now that our system is at an energy minimum, we can begin real dynamics.
EM ensured that we have a reasonable starting structure, in terms of geometry and solvent orientation. To begin real dynamics, we must equilibrate the solvent and ions around the protein. If we were to attempt unrestrained dynamics at this point, the system may collapse. The reason is that the solvent is mostly optimized within itself, and not necessarily with the solute. It needs to be brought to the temperature we wish to simulate and establish the proper orientation about the solute (the protein). After we arrive at the correct temperature (based on kinetic energies), we will apply pressure to the system until it reaches the proper density.
Remember that posre.itp file that pdb2gmx generated a long time ago? We're going to use it now. The purpose of posre.itp is to apply a position restraining force on the heavy atoms of the protein (anything that is not a hydrogen). Movement is permitted, but only after overcoming a substantial energy penalty. The utility of position restraints is that they allow us to equilibrate our solvent around our protein, without the added variable of structural changes in the protein.
Equilibration is often conducted in two phases. The first phase is conducted under an
NVT ensemble (constant Number of particles, Volume, and Temperature). This ensemble is also referred to as "isothermal-isochoric" or "canonical." The timeframe for such a procedure is dependent upon the contents of the system, but in
NVT, the temperature of the system should reach a plateau at the desired value. If the temperature has not yet stabilized, additional time will be required. Typically, 50-100 ps should suffice, and we will conduct a 100-ps
NVT equilibration for this exercise. Depending on your machine, this may take a while (just over an hour on a dual-core MacBook). Get the .mdp file
here.
We will call grompp and mdrun just as we did at the EM step:
grompp -f nvt.mdp -c em.gro -p topol.top -o nvt.tpr
mdrun -deffnm nvt
A full explanation of the parameters used can be found in the GROMACS manual, in addition to the comments provided. Take note of a few parameters in the .mdp file:
- gen_vel = yes: Initiates velocity generation. Using different random seeds (gen_seed) gives different initial velocities, and thus multiple (different) simulations can be conducted from the same starting structure.
- tcoupl = V-rescale: The velocity rescaling thermostat is an improvement upon the Berendsen weak coupling method, which did not reproduce a correct kinetic ensemble.
- pcoupl = no: Pressure coupling is not applied.
Let's analyze the temperature progression, again using g_energy:
g_energy -f nvt.edr









+14.39.37.png)